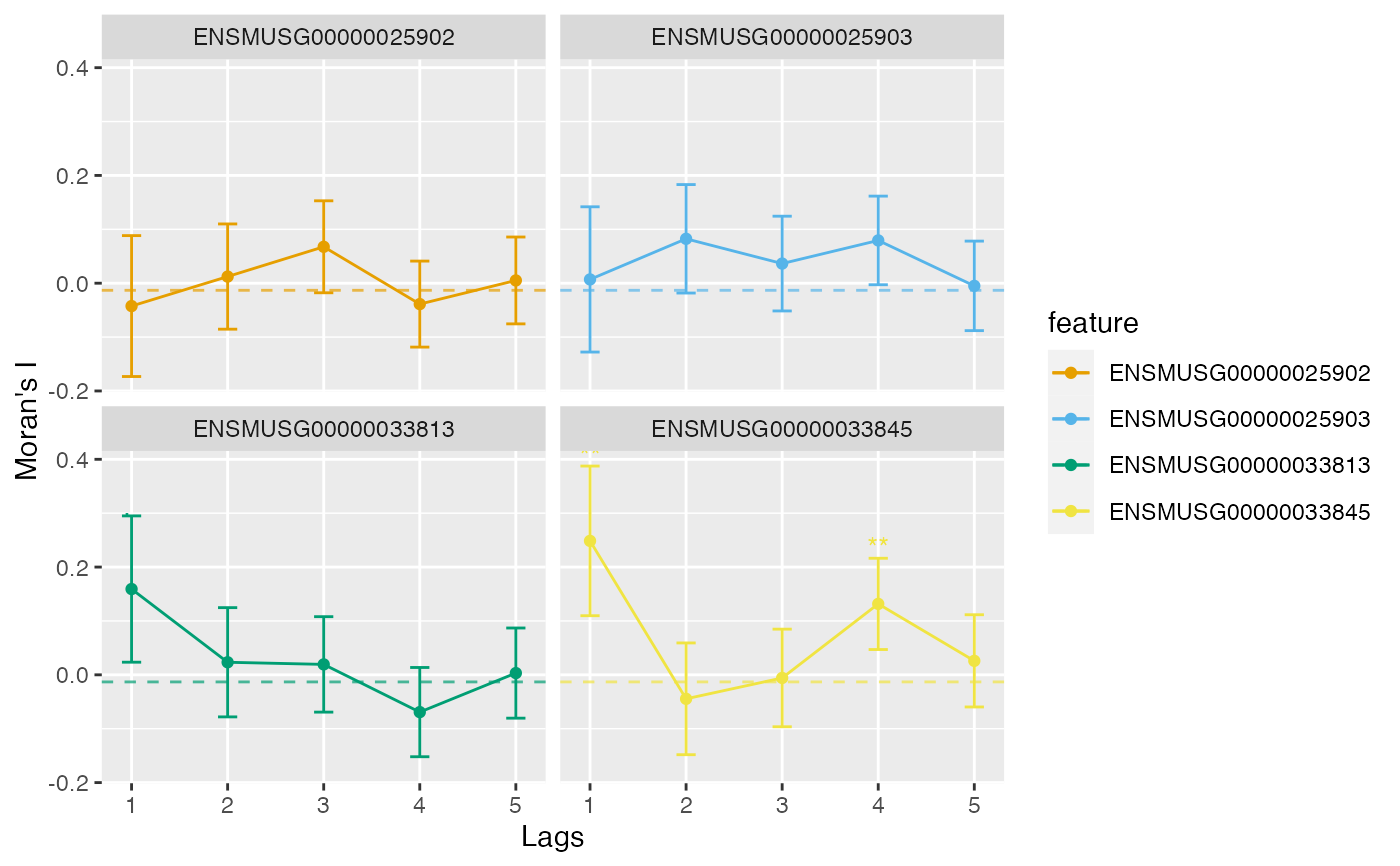
Use ggplot2 to plot correlograms computed by
runUnivariate, pulling results from rowData.
Correlograms of multiple genes with error bars can be plotted, and they can
be colored by any numeric or categorical column in rowData or a vector
with the same length as nrow of the SFE object. The coloring is useful
when the correlograms are clustered to show types of length scales or
patterns of decay of spatial autocorrelation. For method = "I", the
error bars are twice the standard deviation of the estimated Moran's I value.
Usage
plotCorrelogram(
sfe,
features,
sample_id = "all",
method = "I",
color_by = NULL,
facet_by = c("sample_id", "features"),
ncol = NULL,
colGeometryName = NULL,
annotGeometryName = NULL,
reducedDimName = NULL,
plot_signif = TRUE,
p_adj_method = "BH",
divergent = FALSE,
diverge_center = NULL,
swap_rownames = NULL,
name = "sp.correlogram"
)Arguments
- sfe
A
SpatialFeatureExperimentobject.- features
Features to plot, must be in rownames of the gene count matrix, colnames of colData or a colGeometry, colnames of cell embeddings in
reducedDim, or numeric indices of dimension reduction components.- sample_id
Sample(s) in the SFE object whose cells/spots to use. Can be "all" to compute metric for all samples; the metric is computed separately for each sample.
- method
"corr" for correlation, "I" for Moran's I, "C" for Geary's C
- color_by
Name of a column in
rowData(sfe)or in thefeatureDataofcolData(seecolFeatureData),colGeometry, orannotGeometryby which to color the correlogram of each feature. Alternatively, a vector of the same length asfeatures, or a data frame fromclusterCorrelograms.- facet_by
Whether to facet by sample_id (default) or features. If facetting by sample_id, then different features will be plotted in the same facet for comparison. If facetting by features, then different samples will be compared for each feature. Ignored if only one sample is specified.
- ncol
Number of columns if facetting.
- colGeometryName
Name of a
colGeometrysfdata frame whose numeric columns of interest are to be used to compute the metric. UsecolGeometryNamesto look up names of thesfdata frames associated with cells/spots.- annotGeometryName
Name of a
annotGeometryof the SFE object, to annotate the gene expression plot.- reducedDimName
Name of a dimension reduction, can be seen in
reducedDimNames.colGeometryNameandannotGeometryNamehave precedence overreducedDimName.- plot_signif
Logical, whether to plot significance symbols: p < 0.001: ***, p < 0.01: **, p < 0.05 *, p < 0.1: ., otherwise no symbol. The p-values are two sided, based on the assumption that the estimated Moran's I is normally distributed with mean from a randomized version of the data. The mean and variance come from
moran.testfor Moran's I andgeary.testfor Geary's C. Take the results with a grain of salt if the data is not normally distributed.- p_adj_method
Multiple testing correction method as in
p.adjust, to correct for multiple testing (number of lags times number of features) in the Moran's I estimates ifplot_signif = TRUE.- divergent
Logical, whether a divergent palette should be used.
- diverge_center
If
divergent = TRUE, the center from which the palette should diverge. IfNULL, then not centering.- swap_rownames
Column name of
rowData(object)to be used to identify features instead ofrownames(object)when labeling plot elements. If not found inrowData, then rownames of the gene count matrix will be used.- name
Name under which the correlogram results are stored, which is by default "sp.correlogram".
Examples
library(SpatialFeatureExperiment)
library(SFEData)
library(bluster)
library(scater)
sfe <- McKellarMuscleData("small")
#> see ?SFEData and browseVignettes('SFEData') for documentation
#> loading from cache
sfe <- logNormCounts(sfe)
colGraph(sfe, "visium") <- findVisiumGraph(sfe)
inds <- c(1, 3, 4, 5)
features <- rownames(sfe)[inds]
sfe <- runUnivariate(sfe,
type = "sp.correlogram", features = features,
exprs_values = "counts", order = 5
)
clust <- clusterCorrelograms(sfe,
features = features,
BLUSPARAM = KmeansParam(2)
)
# Color by features
plotCorrelogram(sfe, features)
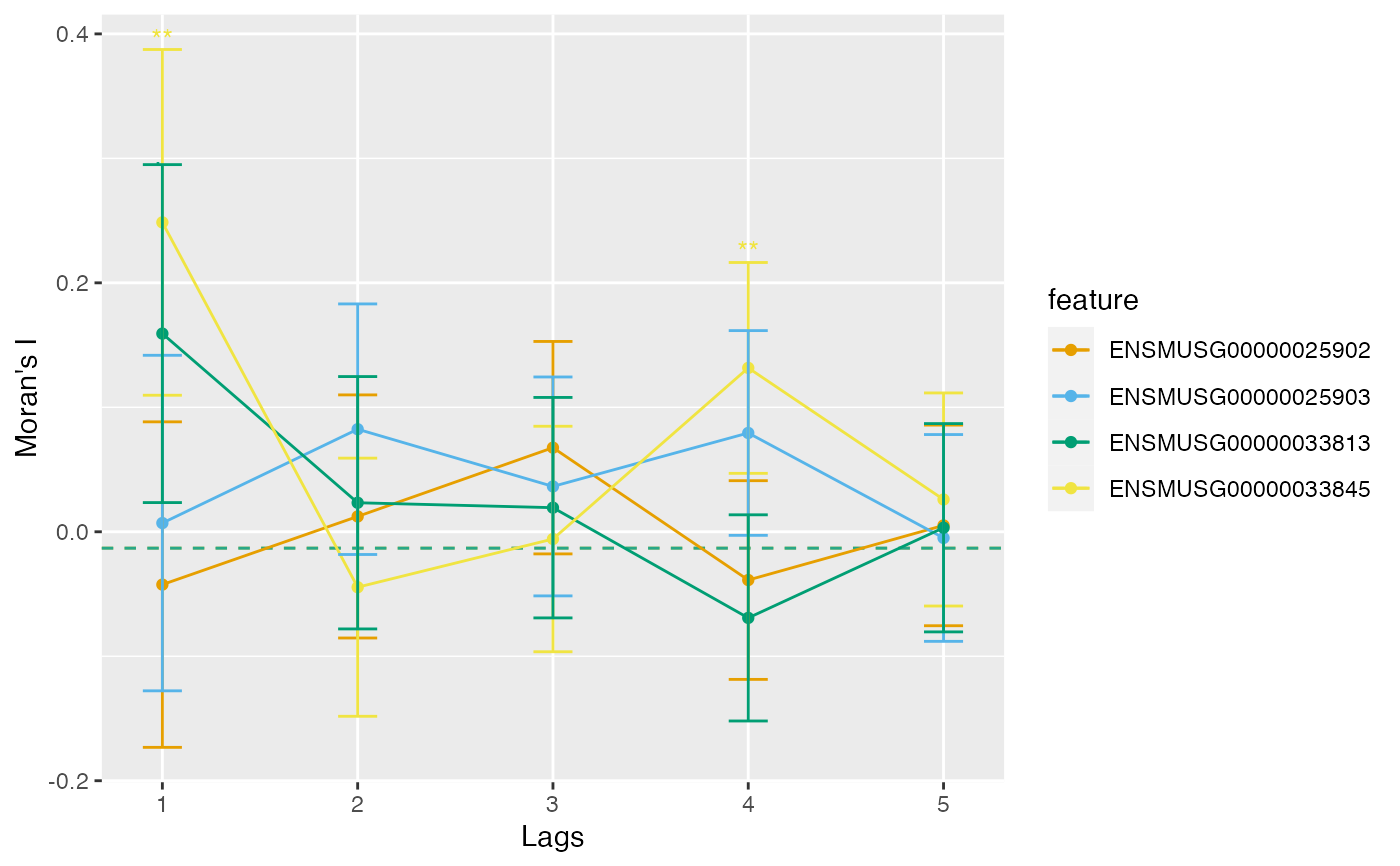 # Color by something else
plotCorrelogram(sfe, features, color_by = clust$cluster)
# Color by something else
plotCorrelogram(sfe, features, color_by = clust$cluster)
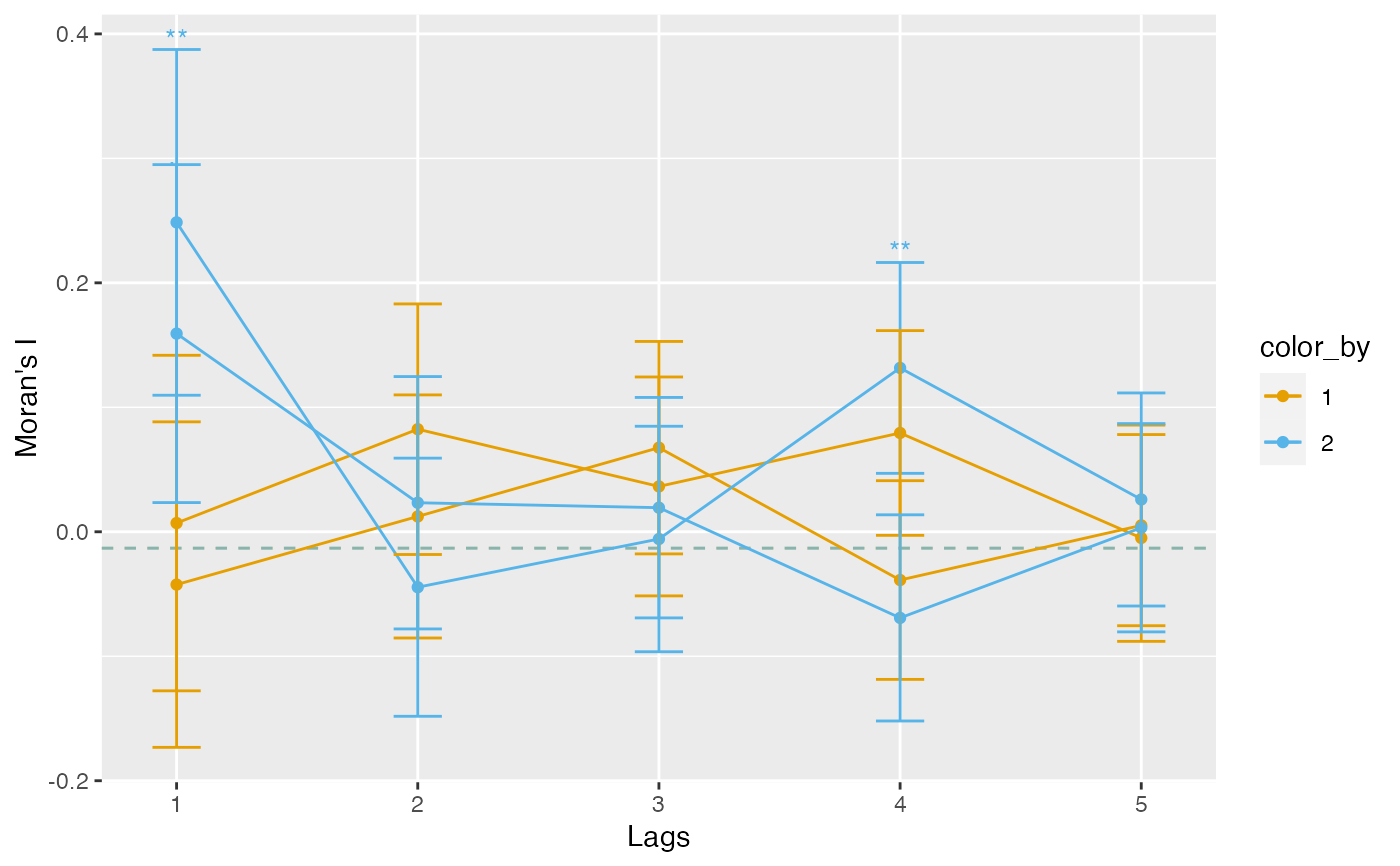 # Facet by features
plotCorrelogram(sfe, features, facet_by = "features")
# Facet by features
plotCorrelogram(sfe, features, facet_by = "features")