Such as plotting the value of projection of gene expression of each cell to a principal component in space. At present, this function does not work for the 3D array of geographically weighted PCA (GWPCA), but a future version will deal with GWPCA results.
Usage
spatialReducedDim(
sfe,
dimred,
ncomponents = NULL,
components = ncomponents,
colGeometryName = 1L,
sample_id = "all",
ncol = NULL,
ncol_sample = NULL,
annotGeometryName = NULL,
rowGeometryName = NULL,
rowGeometryFeatures = NULL,
annot_aes = list(),
annot_fixed = list(),
tx_fixed = list(),
exprs_values = "logcounts",
bbox = NULL,
tx_file = NULL,
image_id = NULL,
channel = NULL,
maxcell = 5e+05,
aes_use = c("fill", "color", "shape", "linetype"),
divergent = FALSE,
diverge_center = NULL,
annot_divergent = FALSE,
annot_diverge_center = NULL,
size = 0,
shape = 16,
linewidth = 0,
linetype = 1,
alpha = 1,
color = NA,
fill = "gray80",
scattermore = FALSE,
pointsize = 0,
bins = NULL,
summary_fun = sum,
hex = FALSE,
show_axes = FALSE,
dark = FALSE,
palette = colorRampPalette(c("black", "white"))(255),
normalize_channels = FALSE,
...
)Arguments
- sfe
A
SpatialFeatureExperimentobject.- dimred
A string or integer scalar indicating the reduced dimension result in
reducedDims(sfe)to plot.- ncomponents
A numeric scalar indicating the number of dimensions to plot, starting from the first dimension. Alternatively, a numeric vector specifying the dimensions to be plotted.
- components
A numeric scalar or vector specifying which dimensions to be plotted. Use this instead of
ncomponentswhen plotting only one dimension.- colGeometryName
Name of a
colGeometrysfdata frame whose numeric columns of interest are to be used to compute the metric. UsecolGeometryNamesto look up names of thesfdata frames associated with cells/spots.- sample_id
Sample(s) in the SFE object whose cells/spots to use. Can be "all" to compute metric for all samples; the metric is computed separately for each sample.
- ncol
Number of columns if plotting multiple features. Defaults to
NULL, which means using the same logic asfacet_wrap, which is used bypatchwork'swrap_plotsby default.- ncol_sample
If plotting multiple samples as facets, how many columns of such facets. This is distinct from
ncols, which is for multiple features. When plotting multiple features for multiple samples, then the result is a multi-panel plot each panel of which is a plot for each feature facetted by samples.- annotGeometryName
Name of a
annotGeometryof the SFE object, to annotate the gene expression plot.- rowGeometryName
Name of a
rowGeometryof the SFE object to plot.- rowGeometryFeatures
Which features from
rowGeometryto plot. Can only be a small number to avoid overplotting. Different features are distinguished by point shape. By default (NULL), whenrowGeometryNameis specified, this will be whichever items infeaturesthat are also in the row names of the SFE object. If features specified for this argument are not the same as or a subset of those in argumentfeatures, then the spots of all features specified here will be plotted, differentiated by point shape.- annot_aes
A named list of plotting parameters for the annotation sf data frame. The names are which geom (as in ggplot2, such as color and fill), and the values are column names in the annotation sf data frame. Tidyeval is NOT supported.
- annot_fixed
Similar to
annot_aes, but for fixed aesthetic settings, such ascolor = "gray". The defaults are the same as the relevant defaults for this function.- tx_fixed
Similar to
annot_fixed, but to specify fixed aesthetic for transcript spots.- exprs_values
Integer scalar or string indicating which assay of x contains the expression values.
- bbox
A bounding box to specify a smaller region to plot, useful when the dataset is large. Can be a named numeric vector with names "xmin", "xmax", "ymin", and "ymax", in any order. If plotting multiple samples, it should be a matrix with sample IDs as column names and "xmin", "ymin", "xmax", and "ymax" as row names. If multiple samples are plotted but
bboxis a vector rather than a matrix, then the same bounding box will be used for all samples. You may see points at the edge of the geometries if the intersection between the bounding box and a geometry happens to be a point there. IfNULL, then the entire tissue is plotted.- tx_file
File path to GeoParquet file of the transcript spots if you don't wish to load all transcript spots into the SFE object. See
formatTxSpotson generating such a GeoParquet file.- image_id
ID of the image to plot behind the geometries. If
NULL, then not plotting images. UseimgDatato see image IDs present. To plot multiple grayscale images as different RGB channels, use a named vector here, whose names are channel names (r, g, b), and values are image_ids of the corresponding images. The RGB colorization may not be colorblind friendly. When plotting multiple samples, it is assumed that the same image_id is used for each channel across different samples.- channel
Numeric vector indicating which channels in a multi-channel image to plot. If
NULL, grayscale is plotted if there is 1 channel and RGB for the first 3 channels. The numeric vector can be named (r, g, b) to indicate which channel maps to which color. The RGB colorization may not be colorblind friendly. This argument cannot be specified whileimage_idis a named vector to plot different grayscale images as different channels.- maxcell
Maximum number of pixels to plot in the image. If the image is larger, it will be resampled so it have less than this number of pixels to save memory and for faster plotting. We recommend reducing this number when plotting multiple facets.
- aes_use
Aesthetic to use for discrete variables. For continuous variables, it's always "fill" for polygons and point shapes 21-25. For discrete variables, it can be fill, color, shape, or linetype, whenever applicable. The specified value will be changed to the applicable equivalent. For example, if the geometry is point but "linetype" is specified, then "shaped" will be used instead.
- divergent
Logical, whether a divergent palette should be used.
- diverge_center
If
divergent = TRUE, the center from which the palette should diverge. IfNULL, then not centering.- annot_divergent
Just as
divergent, but for the annotGeometry in case it's different.- annot_diverge_center
Just as
diverge_center, but for the annotGeometry in case it's different.- size
Fixed size of points. For points defaults to 0.5. Ignored if
size_byis specified.- shape
Fixed shape of points, ignored if
shape_byis specified and applicable.- linewidth
Width of lines, including outlines of polygons. For polygons, this defaults to 0, meaning no outlines.
- linetype
Fixed line type, ignored if
linetype_byis specified and applicable.- alpha
Transparency.
- color
Fixed color for
colGeometryifcolor_byis not specified or not applicable, or forannotGeometryifannot_color_byis not specified or not applicable.- fill
Similar to
color, but for fill.- scattermore
Logical, whether to use the
scattermorepackage to greatly speed up plotting numerous points. Only used for POINTcolGeometries. If the geometry is not POINT, then the centroids are used. Recommended for plotting hundreds of thousands or more cells where the cell polygons can't be seen when plotted due to the large number of cells and small plot size such as when plotting multiple panels for multiple features.- pointsize
Radius of rasterized point in
scattermore. Default to 0 for single pixels (fastest).- bins
If binning the
colGeometryin space due to large number of cells or spots, the number of bins, passed togeom_bin2dorgeom_hex. IfNULL(default), then thecolGeometryis plotted without binning. If binning, a point geometry is recommended. If the geometry is not point, then the centroids will be used.- summary_fun
Function to summarize the feature value when the
colGeometryis binned.- hex
Logical, whether to use
geom_hex. Note thatgeom_hexis broken inggplot2version 3.4.0. Please updateggplot2if you are getting horizontal stripes whenhex = TRUE.- show_axes
Logical, whether to show axes.
- dark
Logical, whether to use dark theme. When using dark theme, the palette will have lighter color represent higher values as if glowing in the dark. This is intended for plotting gene expression on top of fluorescent images.
- palette
Vector of colors to use to color grayscale images.
- normalize_channels
Logical, whether to normalize each channel of the image individually. Should be
FALSEfor bright field color images such as H&E but should set toTRUEfor fluorescent images.- ...
Other arguments passed to
wrap_plots.
Value
Same as in plotSpatialFeature. A ggplot2 object
if plotting one component. A patchwork object if plotting multiple
components.
Examples
library(SFEData)
library(scater)
sfe <- McKellarMuscleData("small")
#> see ?SFEData and browseVignettes('SFEData') for documentation
#> loading from cache
sfe <- logNormCounts(sfe)
sfe <- runPCA(sfe, ncomponents = 2)
spatialReducedDim(sfe, "PCA", ncomponents = 2, "spotPoly",
annotGeometryName = "tissueBoundary",
divergent = TRUE, diverge_center = 0
)
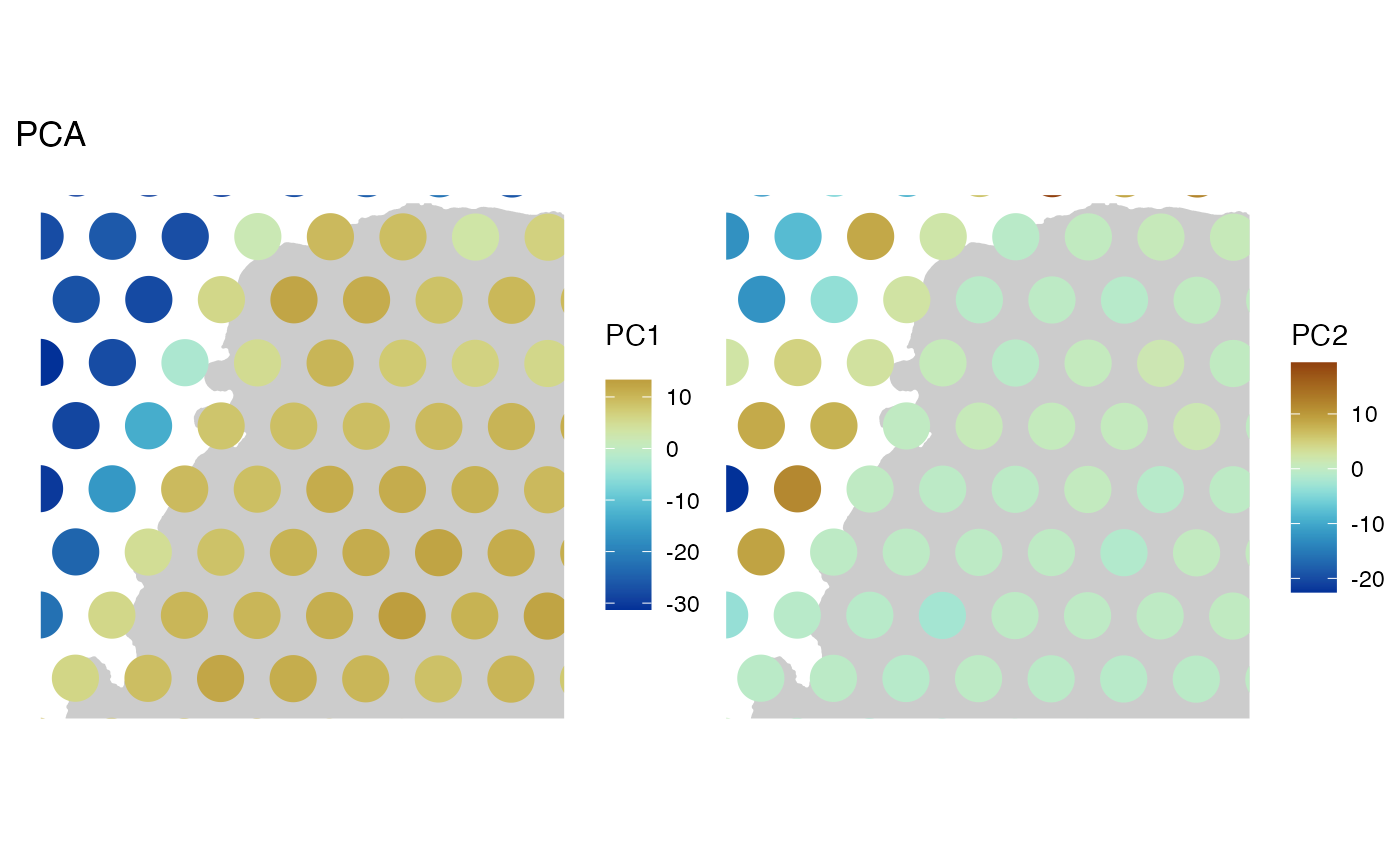 # Basically PC1 separates spots not on tissue from those on tissue.
# Basically PC1 separates spots not on tissue from those on tissue.